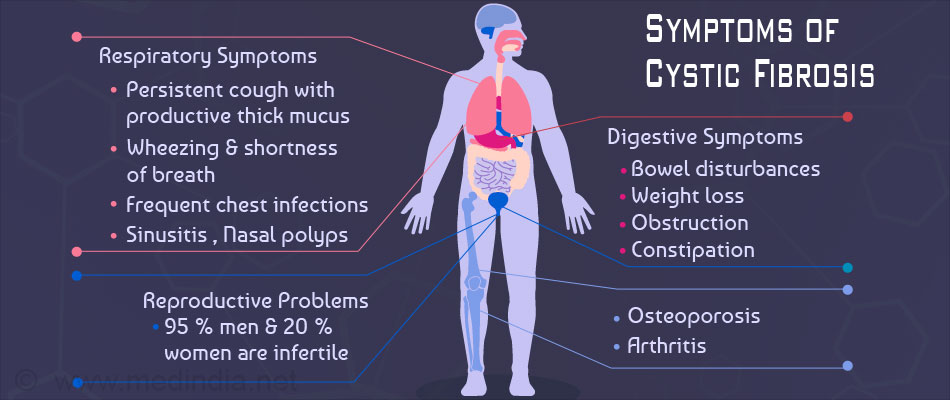
Cystic fibrosis is a genetic disease particularly affecting the children, causing digestive and pulmonary abnormalities. Children who suffer from cystic fibrosis are at increased risk of developing lung infections. This disorder targets the epithelial cells that are present in the inner and luminal lining of the lungs, gastrointestinal tract, pancreas, reproductive organs, and liver.
.jpg)
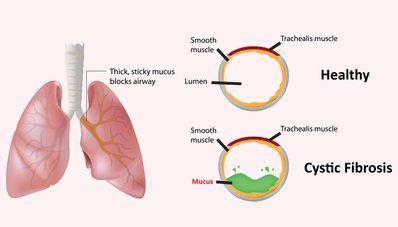
In cystic fibrosis, epithelial cells start to produce an abnormal protein known as cystic fibrosis transmembrane conductance regulator (CFTR) due to some inherited genetic defect. Due to defective protein, transport of chloride found in the cells, and the extracellular matrix is disturbed. Mucus lining inside the airways of the lungs becomes immotile, sticky, and thick as a result of abnormal transport of chloride in the epithelial cells of the lungs. Because of the change in the nature of mucus in the lungs, it fails to remove the trapped pathogens and germs that get infected, causing serious disorders. Electrolytes imbalance disturbs other body systems as well, especially pancreatic and digestive systems.
Due to the dysfunction of the cells of the pancreas, pancreatic ducts are blocked. The blockade of pancreatic ducts does not allow digestive enzymes to reach the digestive tract, which causes GI abnormalities. Abnormal digestion and absorption of food can cause weakness and weight loss in children.
Symptoms:
Children suffering from cystic fibrosis start to show symptoms within one year after birth. The severity and nature of disease vary among different children. In some children, the disease may become quiescent for many years, and they do not show any symptoms. Some babies may show symptoms right after birth, such as meconium ileus. It is a disorder characterized by obstruction of the intestine due to the thickness of the first stool. Symptoms of cystic fibrosis include:
.jpg)
- Excess secretion of salts by the skin glands
- Constant coughing, wheezing, and other hypersensitivity symptoms
- Increased sweating
- Thick bloody mucus
- Fingers and toes deformities
- Frequent sinus and lung infections
- Greasy stools or diarrhea
- Unable to absorb nutrients from the diet
- Increased heart size
- Poor growth and development of children
- Difficulty in breathing during work and rest
- Weight loss
- Rectal prolapse
- Abdominal discomfort and pain
- Nasal polyps that may extend to the pharynx
- Liver disorders
- Diabetes
Risk Factors:
Children that have a family history of cystic fibrosis are at increased risk of developing the disease. Although, cystic fibrosis is a recessive disorder, so children who inherit two copies of this gene usually develop the disease. Children with one copy of defective gene do not suffer from the disease but are carriers that can pass the defective gene to their children.
Patients who are carriers of this recessive disorder do not show any symptoms. If both parents are carriers, there is a 25% chance that their children will develop a symptomatic disorder.
Caucasians in northern Europe are at high risk of developing the disease.
Diagnosis:
Tests are available that can check a fetus with cystic fibrosis during pregnancy. But these investigations are unable to check whether symptoms will be mild or severe.
Newborn screening may be a useful technique to diagnose cystic fibrosis. Newborn screening primarily includes a blood test. If the blood test is positive, your doctor may order a sweat test.
The sweat test is a recommended test for cystic fibrosis because it diagnoses the electrolyte levels in the sweat of a person. This test is usually performed twice. Newborn babies are diagnosed by sweat test after 10 days of birth. If both tests determine the high electrolyte levels, cystic fibrosis is confirmed.
Other tests that can prove helpful in diagnosing the disease or symptoms may include:
- Lung function tests for evaluation of breathing volumes
- Blood tests to determine nutrient levels
- Tests to assess pancreatic functions
- Chest X-rays
- Gram stain and Culture for bacteria in the lungs
Treatment:
.jpg)
The main goals of treatment of cystic fibrosis include decreasing the severity of symptoms, increase lifespan, and reduce complications.
Dietary Modifications:
Children with cystic fibrosis need dietary modifications that include increasing intake of minerals, vitamins, and digestive enzymes. These modifications help to gain weight, increase growth, and prevent deficiencies.
Immunizations:
Children suffering from cystic fibrosis are at high risk of infections. Proper and complete vaccination can help in this case.

Medications:
Cystic fibrosis patients need medication that can lessen inflammation, treat infections, and enhance lung functions. Drugs that are effective in cystic fibrosis include:
- Antibiotics for pulmonary infections
- Anti-inflammatory drugs
- Bronchodilators
- Mucus-thinning medications
- Insulin injections for proper pancreatic functions
Other treatment strategies include:
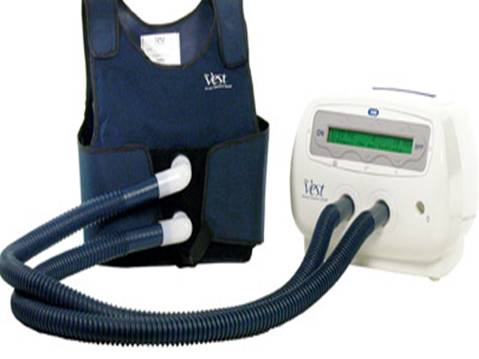
- Chest physical therapy (To loosen and bring up mucus from lungs)
- Surgery (For removing nasal polyps, removing pancreatic and bowel obstruction, liver and lung transplant)
Other medical procedures (Feeding tube, Mucus suction, Oxygen therapy)
